SRAデータを使ったRNA-seqを初めて行ったのでその記録を残しときます。
使ったのは国立遺伝学研究所のスパコン環境で使えるRhelixa RNA-seq解析パイプライン

これをさっそく適当にダウンロードしてきたSRAデータでページに書いてある通り、解析してみました。
$module load singularity/3.5.2
$export GEA_HOME=/lustre7/singularity/images/gene_expression_analysis
$singularity exec -B ${GEA_HOME}/refs:${GEA_HOME}/refs ${GEA_HOME}/gene_expression_analysis.sif GeneExpressionAnalysisSingle.sh Sample hg19 SRR13764564_1.fastq SRR13764564_2.fastq
で動きましたが、、、
[warning][os,thread] Failed to start thread - pthread_create failed (EAGAIN) for attributes: stacksize: 1024k, guardsize: 4k, detached.
というエラーがめっちゃでました。これはスパコンにログインするときにメモリサイズを指定しておかないとデフォルトだと小さいとのことです。
メモリサイズの指定をしなかったからか、なかなか時間がかかりましたがとりあえず出力はえられました。
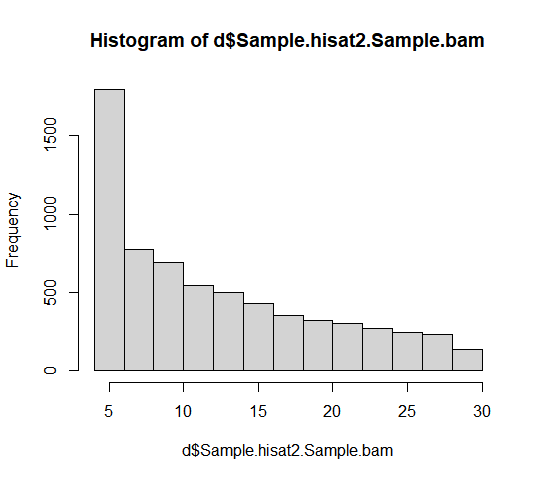
Sample/featureCounts/Sample_count.txtをRで見てみる。
この方の記録を参考にそのまま頑張ってみた。
$breaks
[1] 4 6 8 10 12 14 16 18 20 22 24 26 28 30
$counts
[1] 1792 771 693 545 499 427 351 322 301 269 242 232 133
$density
[1] 0.13623232 0.05861335 0.05268359 0.04143226 0.03793523 0.03246161
[7] 0.02668390 0.02447925 0.02288277 0.02045005 0.01839745 0.01763722
[13] 0.01011099
$mids
[1] 5 7 9 11 13 15 17 19 21 23 25 27 29
$xname
[1] "d$Sample.hisat2.Sample.bam"
$equidist
[1] TRUE
attr(,"class")
[1] "histogram"
ここまでは順調にできた。
☆正規化☆
edgeR か DEseq で作業してみようと思うがその前に勉強が必要そう
インストール手順などはまたこの方のを見ながら勉強させていただいた。
Rhelixa RNA-seq解析パイプラインの問題点は複数のbamファイルをfeatureCountsに入れれなさそうなところ…(勘違いだったらすいません…)
イメージではこの方の紹介しているように全部放り込みたい

けど、まぁ一個ずつのファイルの7カラム目を回収していけば良いっちゃ良いか…
また次回!

コメント